

【ポイント】
- 量子化学計算と微分計算をもとに、新たな触媒設計指針を確立。
- 有機リン配位子のコンピュータ上での高速な最適化を実現。
- 量子化学計算を駆使した新たな反応開発プロセスへの発展に期待。
【概要】
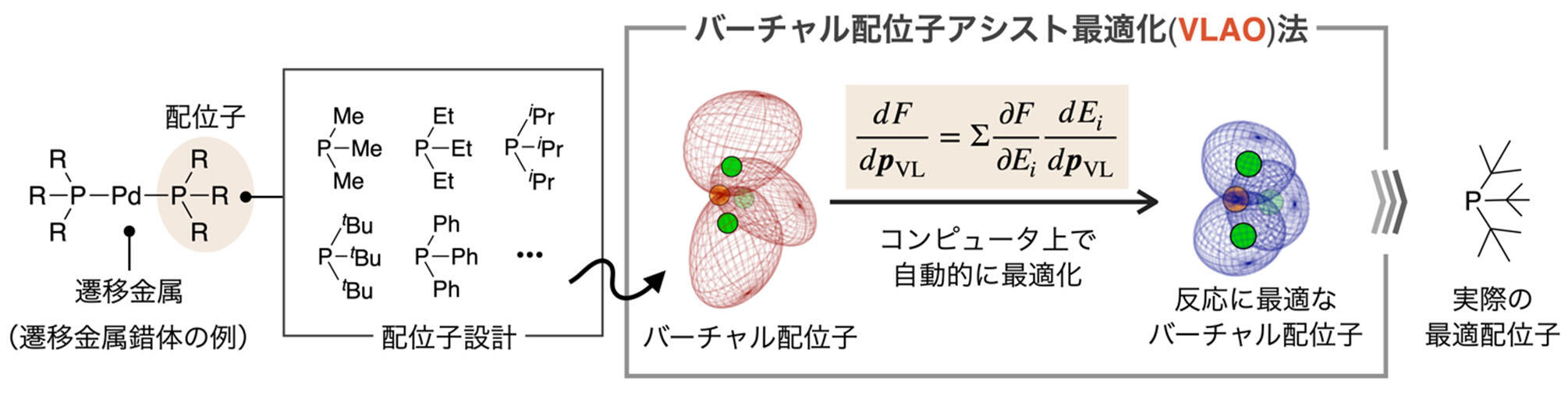
北海道大学創成研究機構化学反応創成研究拠点(WPI-ICReDD)、同大学大学院理学研究院の松岡和助教、前田理教授、WPI-ICReDDの大城泰平特任准教授、WPI-ICReDD及び東京大学大学院情報理工学系研究科の岩田覚教授、英国ブリストル大学のフェイ・ナタリー准教授らの研究グループは、遷移金属触媒反応の開発において最も重要な過程の一つである配位子設計を、量子化学計算によって加速させる「バーチャル配位子アシスト最適化法」を開発しました。
このバーチャル配位子アシスト最適化法は、実験を行うことなくコンピュータ上で最適配位子の予測を可能にするため、従来の触媒設計法において問題であった、開発時間やコストに関する様々な問題を解決し、新しい遷移金属触媒反応開発プロセスへと発展することが期待されます。
なお、本研究成果は、日本時間2024年10月21日(月)公開のACS Catalysisに掲載されました。
論文名:Virtual Ligand-Assisted Optimization: A Rational Strategy for Ligand Engineering(バーチャル配位子アシスト最適化法:配位子設計のための合理的戦略)
URL:https://doi.org/10.1021/acscatal.4c06003
詳細は理学研究院>研究ニュースをご覧ください。