量子化学計算で、有機化合物の出発原料をゼロから予測~網羅的な逆合成解析により高収率な化学反応を予測~
【ポイント】
- 有機化学の知識や実験データを一切使用しない、量子化学計算のみによる逆合成解析を確立。
- ペリ環状反応由来の天然有機化合物に対して逆合成解析を適用し、実験で報告されている出発原料へ立体特異的に逆合成されることを証明。
- 逆合成解析による出発原料の高精度予測は、高収率な化学反応創出技術として有機合成化学分野の発展に大きく貢献することが期待。
【概要】
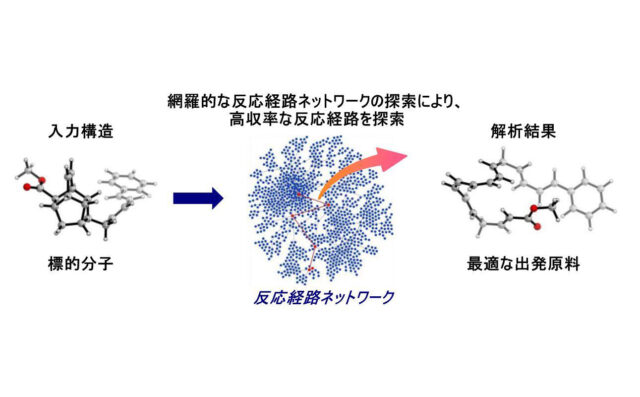
JST事業の1つであるERATO前田化学反応創成知能プロジェクトにおいて、北海道大学創成研究機構化学反応創成研究拠点(WPI-ICReDD)の美多 剛特任准教授、及び同拠点拠点長・同大学院理学研究院の前田 理教授らの研究グループは、ペリ環状反応で得られる生成物(標的分子)を入力構造とし、理化学研究所(理研)のスーパーコンピュータ「富岳」、及び北海道大学の「グランシャリオ」を用いて量子化学的逆合成解析(QCaRA)を実施し、出発原料を正確に予測することに成功しました。
量子化学計算の究極の目的の1つに、反応中間体や遷移状態のエネルギーの計算値を基に『新しい化学反応をゼロから予測し、実験的に実現する』ことが挙げられます。この予測を可能とするためには、可能性のある反応経路を正確にかつ漏れなく計算することによって、反応経路が網目状につながれた「反応経路ネットワーク(反応中間体、及び遷移状態が内包されている)」を構築し、得られた反応経路ネットワーク上で反応物を起点とした速度論シミュレーションを解くことで生成物及び副生成物の反応収率を予想することが求められます。このような解析は、WPI-ICReDDの前田拠点長により開発された反応経路自動探索法である人工力誘起反応法(AFIR法)と速度定数行列縮約法(RCMC法)を組み合わせることで可能となります。また、これらの方法を応用することで、標的分子を入力とした逆探索による化学反応を予測する量子化学的逆合成解析が可能になりました。これにより、有機化学の知識と膨大な実験データに頼ることなく、新しい化学反応をコンピュータ上で予測することができるようになりました。
本研究では、このコンピュータによる反応予測の端緒を担うべく、基本的なペリ環状反応生成物を25個選択し、それに対して逆合成化学的なAFIR法を適用しました。その結果、標的分子の分子構造を1つだけ入力するのみで、実験的に報告されている経路、かつ立体特異性に従い合成の出発点となる出発原料に逆合成されることを証明しました。標的分子の原子数が大きくなると計算時間も増大しますが、52原子ある天然有機化合物の逆合成解析にも成功し、反応予測の足掛かりを築くことができました。
本研究で実施したQCaRAによる出発原料を正確に予測する手法は、次世代型の化学反応創出技術として有機合成化学分野の発展に大きく貢献することが期待されます。
本研究成果は、2022年11月30日(水)公開のJournal of the American Chemical Society誌のオンライン版にArticleとして掲載されました 。
論文名:Prediction of High-Yielding Single-Step or Cascade Pericyclic Reactions for the Synthesis of Complex Synthetic Targets
URL:https://doi.org/10.1021/jacs.2c09830
詳細はプレスリリースをご覧下さい。